-
-
From mouse to human: Cellular morphometric subtype learned from mouse mammary tumors provides prognostic value in human breast cancer
Submitted by Hang Chang on Sat, 11/20/2021 - 19:09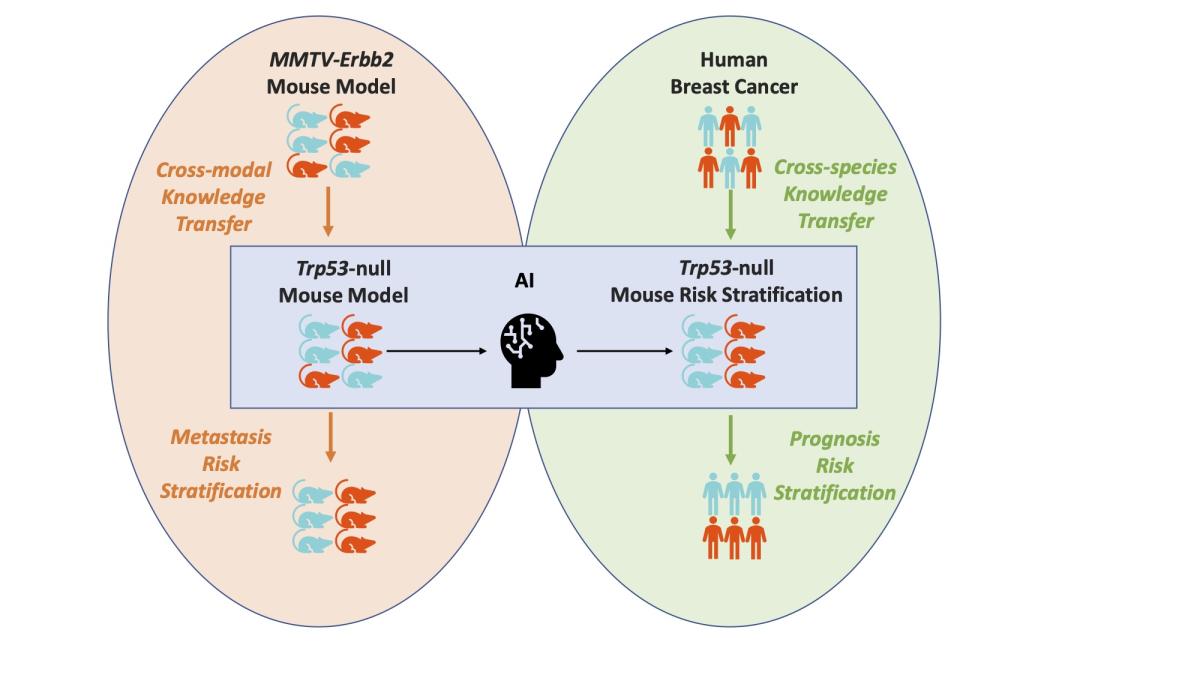
Breast cancer is the second deadliest cancer for women in the United States. How the malignant neoplasm develops, and the treatment necessary to combat the disease, is often as idiosyncratic as the individual affected.
-
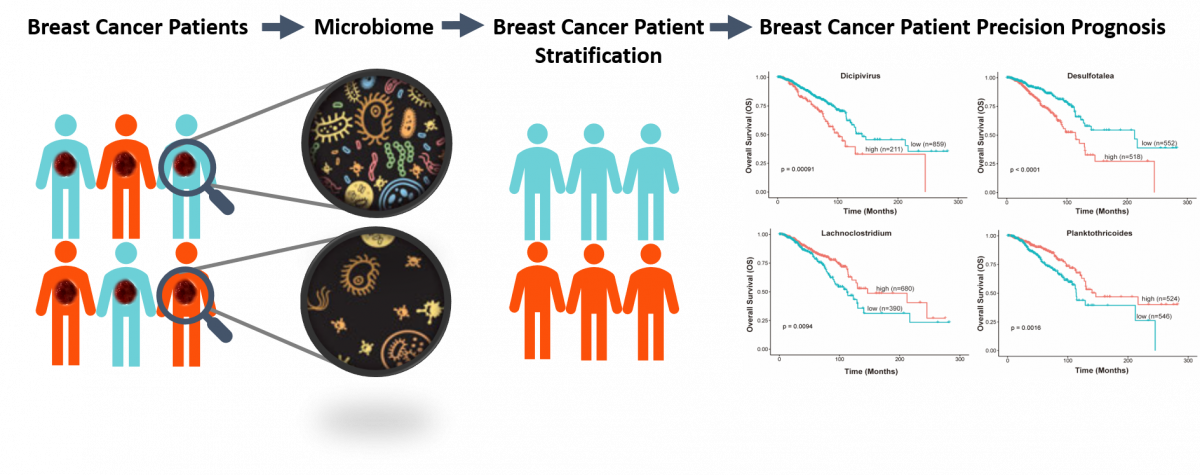
Identification of a novel cancer microbiome signature for predicting prognosis of human breast cancer patients
Submitted by Hang Chang on Thu, 05/27/2021 - 15:27Prognosis of breast cancer (BC) patients differs considerably. Therefore, identify a reliable prognostic biomarker to distinguish patients with poor prognosis. Increasing evidence shows that the microbiome plays a critical role in cancer development and progression, and therapies. Moreover, with the development of microbiome technologies, researchers have revealed the variation in the abundance and composition of intratumor microbiome. In this project, we sought to identify a microbiome signature for predicting the prognosis of patients with breast cancer. This study will provide a new avenue for developing biomarkers for personalized medicine by incorporating cancer microbiome.
-
Cellular Morphometric Subtypes in Lower Grade Gliomas discovered by Machine Learning
Submitted by Hang Chang on Tue, 03/16/2021 - 17:33 -
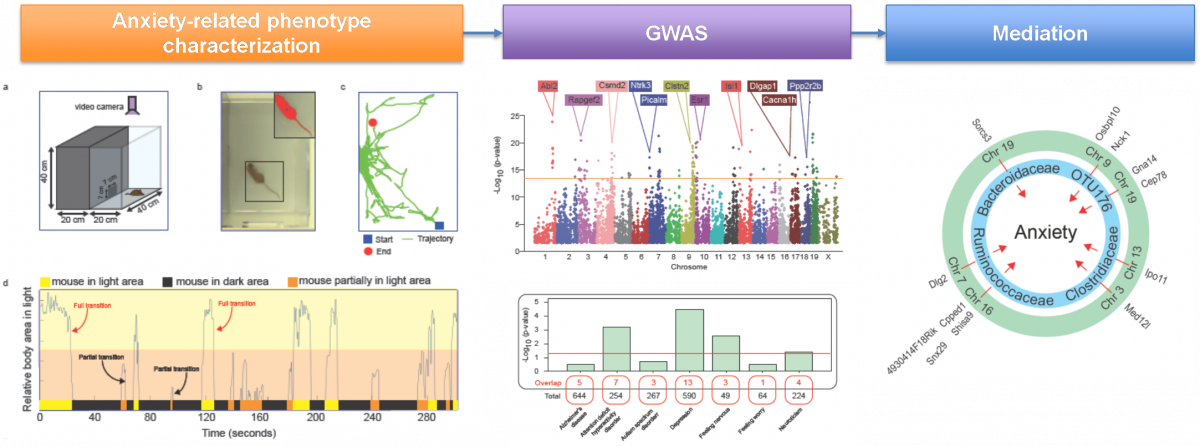
Gut microbiome partially mediates and coordinates the effects of genetics on anxiety-like behavior in Collaborative Cross mice
Submitted by Hang Chang on Mon, 07/27/2020 - 12:56Growing evidence suggests that the gut microbiome (GM) plays a critical role in health and disease. However, the contribution of GM to psychiatric disorders, especially anxiety, remains unclear. We used the Collaborative Cross (CC) mouse population-based model to identify anxiety associated host genetic and GM factors.
Schematic pipeline for our study is illustrated as follows,
-
Multi-modality Informatics Approach for Early Stage Cancer Diagnosis
Submitted by Hang Chang on Fri, 03/17/2017 - 12:58Breast cancer is the most commonly diagnosed cancer in women, and is the second leading cause of cancer death among women in U.S. However, current screening methods for early detection of breast cancer often lead to over-diagnosis and subsequent overtreatment, and recent studies estimated that breast cancer was over-diagnosed in 1.3 million U.S. women in the past 30 years. This research project aims at addressing the overarching challenges of distinguishing deadly from indolent breast cancers, which is a critical step towards conquering the problems of over-diagnosis and over-treatment.
In this project, we aim to develop an advanced multi-modality informatics system by integrating the unique and complementary clinical values, provided by tissue histology sections and state-of-the-art omics assays. The expected outcome is a highly generic, efficient and effective multi-modality informatics system for distinguishing deadly from indolent breast cancers.
-
Development of new mouse models for human cancer
Submitted by Jian-Hua Mao on Thu, 03/17/2016 - 12:54Sporadic tumors, which account for the majority of all human cancers, evolve as the result of a step-wise accumulation of genetic alterations resulting in uncontrolled cell proliferation and a lack of response to apoptotic cues. Such genetic alterations include point mutations, deletions, duplication/amplification, and translocations and these alterations can lead to the enhanced or decreased activity of the expressed protein. These alterations are referred to as ‘gain-of-function’ or ‘loss-of-function’ mutations, respectively. The a??ected genes are termed oncogenes or tumor suppressors, respectively. Within the last decade, the availability of a complete sequence-based map of the human genome, coupled with significance technological advances, has revolutionized the search for somatic alterations in tumor genomes. Within a given tumor type there are many infrequently mutated genes and a few frequently mutated genes, resulting in incredible genetic heterogeneity. The resulting catalogues of somatic alterations will point to candidate cancer genes, but requiring further validation to determine whether they have a causal role in tumorigenesis. The availability of gene targeting and transgenic technology in the mouse gives us unparalleled opportunities to test the functional significance of genetic changes in tumor development. Another one of the broad and long-term goals of my laboratory is to develop new mouse models for human cancer. These mouse models not only will increase our understanding of genetic aberration associated with cancer progression, but also will potentially help to identify personalized medicine for cancer patients, which may eventually contribute to a decrease in morbidity and mortality of cancer.
-
Big data oriented Imaging Bio-Markers identification towards Personalized Therapy
Submitted by Hang Chang on Fri, 02/26/2016 - 18:19Adult primary brain tumors such as gliomas are characterized by enormous cellular diversity captured by grading. For gliomas, tumor grade is based on the region with the highest level of aberrant histopathology. Recently, a large number of subtypes have been characterized based on morphological variants and their molecular characterization showing an enormous heterogeneity that is only being discovered. This poses an enormous challenge in interpreting these subtypes and understanding their clinical and molecular associations.This project aims to develop open source advanced image-based modeling algorithms and software and to couple them with a bioinformatics system for the analysis of brain tumors. The net results are: a more robust identification tumor subtypes; hypothesis generation for the molecular basis of each subtype; creation of a publicly available databank where new tissue sections can be compared against an existing database of prognostic and predictive subtypes and their molecular signatures; and potentially enabling new opportunities for personalized therapy.